背景简介
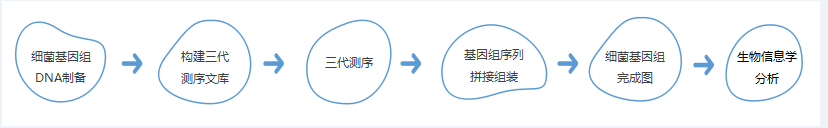
细菌全基因组具有相对较小、重复序列较高、易于突变等特点,通过全基因组测序,可以对细菌基因组进行测序、拼接、组装,获得完整细菌基因信息。细菌de novo测序已取代传统方法成为研究细菌进化遗传机制、关键功能基因的重要工具。目前,细菌全基因组测序利用三代测序结合二代测序的方法可以得到0 Gap的完整的基因组序列;对其进行功能基因注释以及个性化分析,全面解析细菌生物学意义。
技术优势
实验安排灵活、服务周期短、速度快
测序结果准确性更高、成本更低
丰富的项目经验与专业的生信分析团队,提供最全面准确的信息分析
技术路线
分析内容

样本类型
DNA送样:
DNA浓度 ≥20ng/μl(Qubit),DNA质量≥15ug(Qubit)DNA电泳条带单一,无明显降解。
菌体送样:
收集生长对数期菌体,收集离心菌体数3×1010个,于无菌离心管中,液氮速冻,干冰运输
Q1:为何完成图选择三代测序平台?
A:受测序片段长度的限制,细菌基因组序列通常需要利用软件算法将大量测序片段拼接起来,而细菌基因组中重复序列的存在,则会大大增加拼接的复杂度。细菌重复序列的大小从几百bp到7 Kb不等,细菌框架图的插入片段,只能解决少量的重复片段问题,因此组装结果更加碎片化;细菌精细图采用了6 Kb大片段文库,可以跨过绝大部分重复序列,并将结果Scaffold控制在30条以内;而三代测序采用了10 Kb文库,平均读长也达到10 Kb以上,由于序列够长,避免了细菌基因组中重复序列的影响,因此能够获得0 gap的完整组装结果。
Q2:对于细菌基因组测序,三代和二代测序相比有何优势?
A:三代测序相比二代测序而言,其优势在于读长长,GC含量影响小,而劣势是测序成本偏高。对于细菌基因组测序来说,三代测序的长读长可以解决细菌中的重复序列问题,也避免了异常GC菌株的测序不均匀问题。由于细菌基因组较小,需要的测序量不大,对于较为精细的细菌完成图来说,三代成本甚至低于二代结合一代的策略。目前为止,在需要组装完整性较低的细菌框架图层面,二代测序仍能保持一定成本优势。随着三代测序通量提升和成本降低,未来三代测序有望在细菌基因组领域获得更广泛的应用。
Q3:细菌基因组中如何预测核糖体rDNA基因?
A:预测细菌基因组中的核糖体rDNA基因,通常有两种方法:一是通过rDNA序列结构特征进行de novo 预测,二是利用近缘rDNA序列进行同源预测。其中前者预测更准确,但是需要组装结果中具备完整的rDNA结构。在框架图和部分精细图组装结果中,可能有rDNA区域组装不完整,分布于多条scaffold中的情况,会导致de novo 测序方法rDNA预测不到的情况。如果想要获得更完整的预测结果,可以预先提供近缘rDNA序列,使用同源预测方法,以改善预测效果。
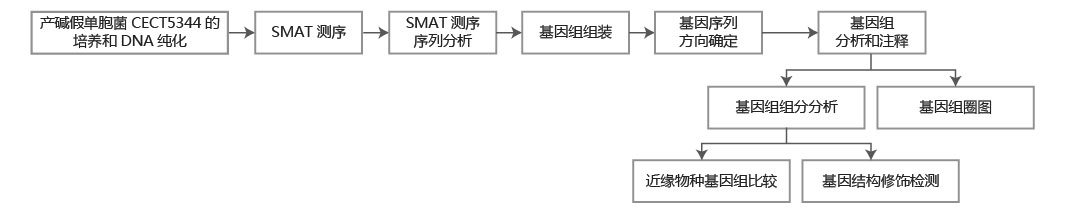
通过单分子实时测序解析可降解氰化物的产碱假单胞菌CECT5344基因组完成图和甲基化情况
研究背景
产碱假单胞杆菌CECT5344在耐受氰化物的同时,还可以在碱性条件下利用氰化物和氰基衍生物作为氮源,极可能作为含氰液体废液污染的生境的生物修复菌。之前已经有该菌株的基因组序列信息,现在采用单分子实时监测序列技术(SMAT)对其基因组进行重测序,得到由GC含量为62.34 %,长4696,984 bp的完整基因组序列。重测序得到的基因组补充了原来基因组中遗漏的部分片段信息,这些遗漏的片段多为转座因子,此外还发现了预测在亚砜还原中起作用的5个基因。CECT5344的基因序列与门多萨假单胞菌高度同源,两者约有70%的基因是相同的。与门多萨假单胞菌不同,CECT5344中并没有发现推断的致病性基因。CECT5344拥有氰水解酶和汞抗性蛋白的独特基因,这些对被氰基和汞化合物污染的环境缓解尤为重要。通过SMAT测序还可以得到菌株的m6A类型的甲基化信息。菌株CECT5344的完整基因组序列为生物学遗传特征的研究提供了基础。
方法流程
研究结果
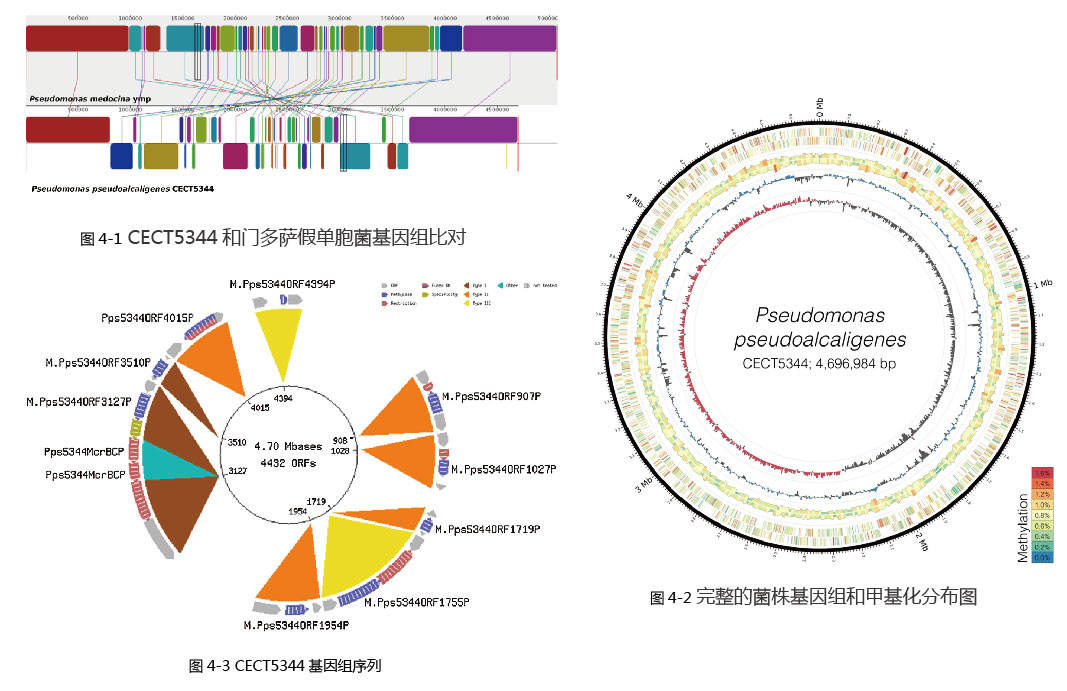
对CECT5344和门多萨假单胞菌基因组比对,对两者的基因结构关系进行了研究,图 4-1中用线连接的色块表示两个基因组的同源区域,最下方的色块代表门多萨假单胞菌基因组中与CECT5344基因组反向的区段。
图 4-2为完整的菌株基因组和甲基化碱基分布图,基因组由4,696,984个碱基对和4436个预测的编码序列组成。圆圈从内到外分别代表:GC偏斜、GC含量、50 kb窗口分析的全基因组甲基化、每个基因的链特异性甲基化、每百万碱基对甲基化的量。
使用REBASE数据库寻找到CECT5344基因组中编码甲基转移酶的基因,共鉴定预测了9个限制/修饰基因的基因座。图 4-3中颜色代码表示不同的限制/修饰类型,蓝色的为甲基化酶,红色为限制性酶。最内层为菌株的基因组完整图和开放阅读框的数目。
参考文献
Daniel Wibberga, Andreas Bremgesb, Tanja Dammann-Kalinowskia,et al.Finished genome sequence and methylome of the cyanide-degradingPseudomonas pseudoalcaligenes strain CECT5344 as resolved bysingle-molecule real-time-sequencing.Journal of Biotechnology, 2016, 232:61-68.
原始数据碱基组成分布例图横坐标是reads 碱基坐标,纵坐标是所有reads 的A、C、 G、T、N 碱基分别占的百分比。每个位置上,A、C、G、 T在开始有所波动,后面会趋于稳定。一般情况下A 与T 相 等,C与G相等,各碱基所占百分比会因物种差异而不同。 基因组项目中,建库比较均匀的情况下,代表不同碱基的 四种颜色的分界线应该波动极小。
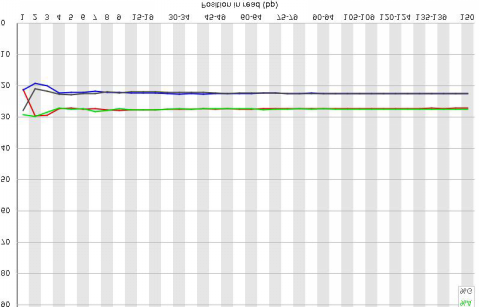
原始数据碱基质量分布例图横坐标是reads 碱基坐标,纵坐标是reads 的碱基质量(SolexaScale: 40=Highest, - 15=Lowest),图中垂直红线”Ⅰ”指定的范围是所有reads 碱基的综合质量,红色垂直方块是质量的四分位值范围,加黑粗线是质量值的中位数。

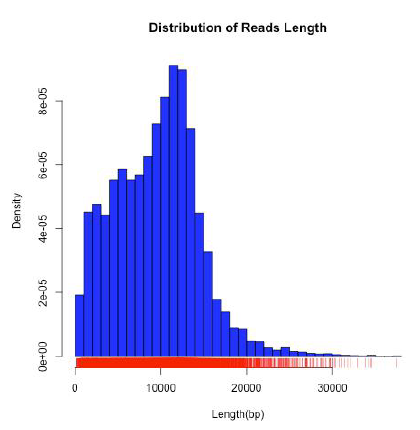
单分子Clean 数据序列的长度分布统计图横坐标为测序reads 的长度,纵坐标为不同长度reads的数目,从上图中可以看出,本次测序获得的reads的长度大小主要集中分布在5000-15000bp,测序质量较高。