背景简介
扩增子测序是对特定长度的PCR产物或捕获的片段进行测序,分析序列中的变异。16S/ITS等扩增子测序即通过提取环境样品的DNA,选择合适的通用引物扩 16S/ITS的目标区域,通过检测目标区域的序列变异和丰度,以研究环境微生物多样性及群落组成差异。16S rDNA为编码原/真核生物核糖体小亚基rRNA的DNA序列。ITS分为两个区域:ITS1位于真核生物rDNA序列18S和5.8S之间,ITS2位于5.8S和28S之间。
技术优势
鉴定到“种”:菌群多样性鉴定率先精细到“种”,分类更明确。
数据库丰富:基于最新版Greengene和自建数据库,和自主开发的分析注释工具,最多可鉴定4414个种,
覆盖2106个属,可鉴定菌种持续更新中。
低成本:相比于传统菌落鉴定,分析通量更高,检测成本更低。
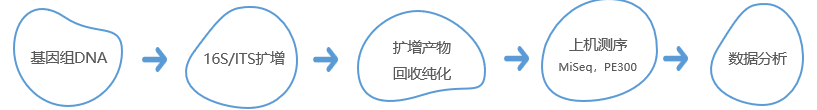
技术路线
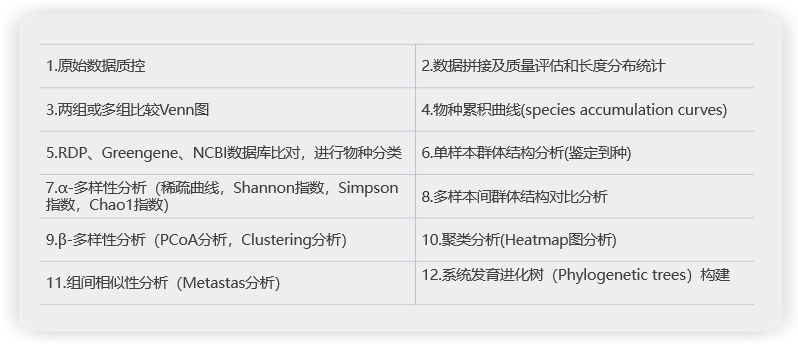
分析内容
样本类型
菌体,DNA等
建议总DNA起始量:>20ng(较纯DNA,无宿主及其他杂质污染)
近期用户文章
1. Gao, S., et al.(2015)Tolerance response to in situ ammonia stress in a pilot-scale anaerobic digestion reactor for alleviating ammonia inhibition. Bioresource Technology 198:372.
2. Gou, H., et al. (2016) Assessment of microbial communities in PM1 and PM10 of Urumqi during winter." Environmental Pollution 214: 202-210.
3. Huang, Y., B. Yang, and W. Li. (2016) Defining the normal core microbiome of conjunctival microbial communities. Clinical Microbiology & Infection 22.7: 643.e7-643.e12.
4. Lv, Long‐Xian, et al. (2016) Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environmental Microbiology 18.7:2272.
5. Hu, Jinxiang, et al. (2016) Pepino (Solanum muricatum) planting increased diversity and abundance of bacterial communities in karst area. Scientific Reports 6:21938.
Q1:什么是嵌合体?
A:嵌合体的形成:在PCR时,当不完全的DNA链与不同的模板退火时,嵌合扩增子形成,并引发衍生自两种不同生物学序列的新模板的合成。嵌合体在正常生物体中是不存在的。参考文献:Robert C. Edgar, UCHIME2: improved chimera prediction for amplicon sequencing,2016.
Q2:PCoA分析有多种不同计算方法及结果,甚至还有其他不同的分析方法来计算样本间距离,我要选哪一种为最终的展示结果呢?
A:PCoA分析和其他不同的分析方法都是用来展示组间差异和组内相似性的结果,不同的计算方法相当于是不同的角度来看这个结果,老师只要选择与实验设计最合适的结果进行展示说明即可。
Q3:α多样性指数之间有什么区别吗?
A:observed_species、chao1指数用来描述物种的数目;而shannon、simpson指数则是用来描述物种多样性的,这两个指数不仅考虑了物种的数目,还考虑了物种的丰度,也就是所有物种的均匀度。如果每一个体都属于不同的种,多样性指数就最大;如果每一个体都属于同一种,则其多样性指数就最小。如果一个样品物种数目很多,但均匀度很差,即某个物种丰度很高,但另一物种丰度很低,就会造成observed_species、chao1指数高而shannon、simpson指数不高的现象。简单说: observed_species、chao1指数高,说明样品物种数目多;shannon、simpson指数高,说明物种丰度以及均匀度都很高。
Q4:微生物群落研究方法的区别?
A:16S rDNA测序:由于研究对象只为细菌的16S rDNA,因此16S测序技术更多只用于研究群落物种信息,也就是利用OTU物种分类、α和β多样性分析等手段解答群落有什么物种,物种关系是什么等问题。因此,这种技术更多地只能了解到环境对微生物的组成有何影响,更偏向于单向关系研究。
宏基因组测序:宏基因组的研究对象为群落所有DNA,因此研究范围更广。理论上不单只可以了解群落的组成和多样性等物种信息,同时,利用基因的注释信息,还可以挖掘群落的核心功能和通路信息,在基因组层面了解这个群落到底有什么物种,这些物种能够发挥什么功能。只有了解群落功能,才能知道它们对环境有什么影响,这有利于进行微生物与环境的双向研究。
宏转录组测序:宏转录组以mRNA为研究对象,同样的通过数据组装和比对,能同时挖掘物种信息,基因功能信息,发现新基因,这与宏基因组的作用没太大差别。它的特点在于,因为是转录组信息,因此在进行基因研究的时候,可以关注到基因表达情况,从而更深入地了解基因如何被调控,基因表达如何应答环境变化等细节问题,在功能研究上更为细致。
定义正常人类眼结膜微生物群落的 "core microbiome"
研究背景
眼部细菌感染是很常见的,但运用传统培养与分子生物学方法鉴定结膜微生物群具有明显的局限性。而宏基因组研究可以弥补这些方法的缺陷。
研究结果
黄钰森组运用Illumina高通量测序技术(MiSeq 测序平台)对结膜擦拭样本中所有细菌的16S rDNA V3-V4高变区进行测序。从测序数据中获得操作分类单元(OTUs)。接着,进行微生物分类、丰度、 α多样性等生物信息学分析。从31个结膜样本中得到840373个高质量测序reads。不同种类的OTU数量从159到2042,显示出很高的微生物多样性。这些细菌菌落可分为25个门和526个不同的属。在属的水平,棒状杆菌属(28.22%)、假单胞菌属(26.75%)、葡萄球菌属(5.28%)、不动杆菌属(4.74%)、链球菌属(2.85%)、 Millisia(2.16%)、厌氧球菌属(1.86%)、大芬戈尔德菌属(1.68%)、西蒙斯氏菌属(1.48%)、韦荣氏球菌属(1.00%)占整个微生物群落的76%以上,可能代表了正常结膜微生物群的“core genera” 。
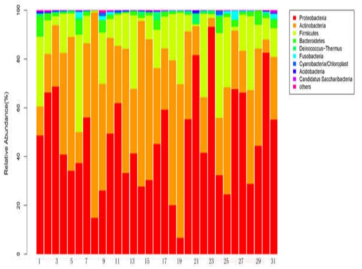
图 眼结膜微生物群落分类
参考文献
Huang YS, et al. (2016) Defining the normal "core microbiome" of conjunctival microbial communities. Clinical Microbiology and Infection. doi:10.1016/j.cmi.2016.04.008.
样品丰度柱状图根据物种丰度表和物种注释表,默认选取丰度最高的20个物种分类,进行相对丰度计算,获得相对丰度文件,绘制样品丰度比较的柱状图,该柱状图以堆叠柱状图 (stacked bar chart)形式展现,便于更直观地进行样品丰度的比较。在各个层级中, 可以直观的看到优势菌种的表达情况及在各个不同处理中的变化趋势。当然, 若是有关注稀有菌群的表达情况时,也可以展现所有物种分类。

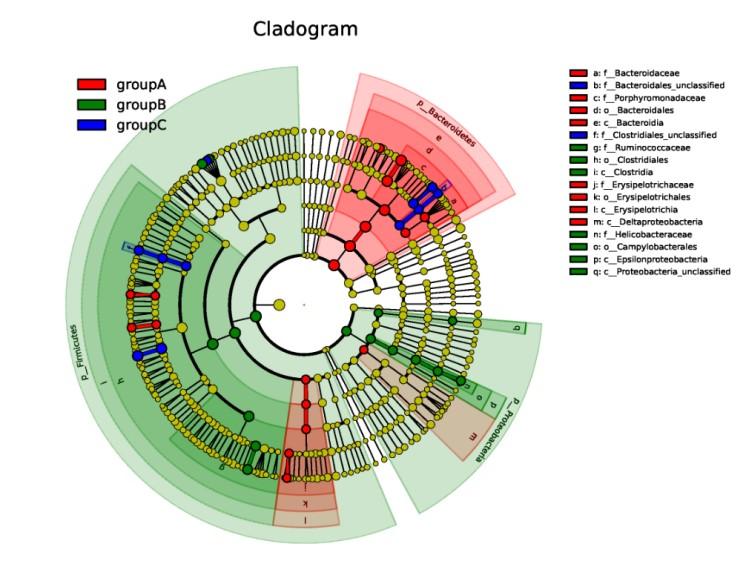
物种分类热图(taxa heatmap)根据样品相对丰度表,将各分类水平相对丰度最高20个的群落组成数据根据分类 单元的丰度分布或样本间的相似程度加以聚类,根据聚类结果对分类单元和样本 分别排序,并通过热图加以呈现。通过聚类,可以将高丰度和低丰度的分类单元 加以区分,并以颜色梯度及相似程度来反映多个样品在各分类水平上组成的相似 性和差异性。
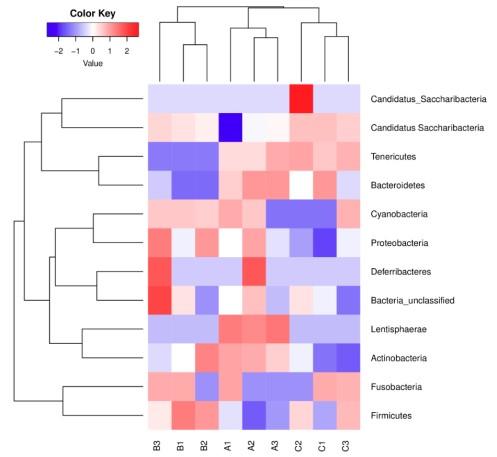
物种注释结果KRONA展示使用Krona软件动态可视化展示单样品在不同分类水平注释结果,通过调节不同的参数来调整展示的图片。如果项目比较关注某一个物种,可通过在Search栏中输入关注物种的名称,可快速定位到关注物种在样品中的表达情况。通过点击左侧栏中的样品名,来展示该样品的物种注释表达情况。
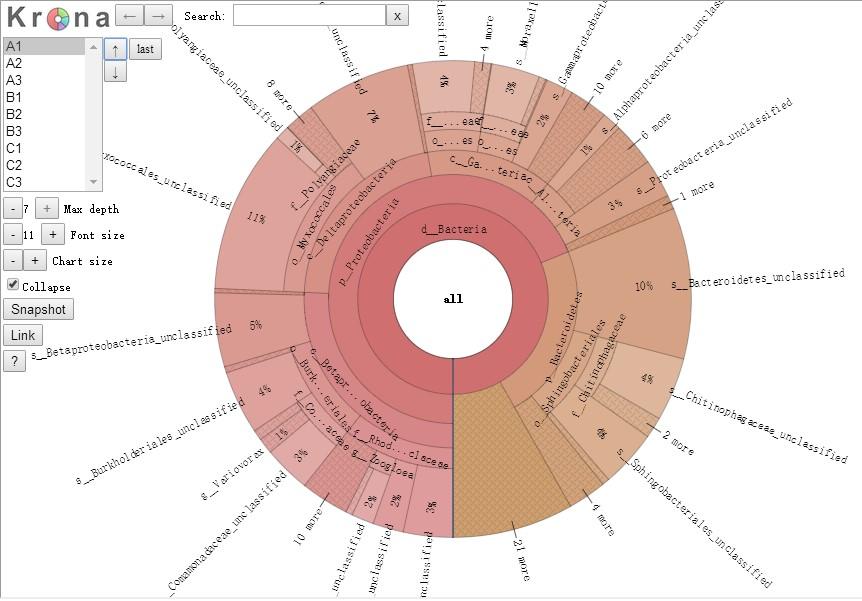
RDA分析RDA分析实际上是约束化的主成分分析(PCA),它的优点是考虑了环境因子(如土壤研究中的PH值,酸碱度,疾病研究中临床理化因子等)对样本的影响, 可同时反映样本,环境因子和物种三者或两两之间的关系。
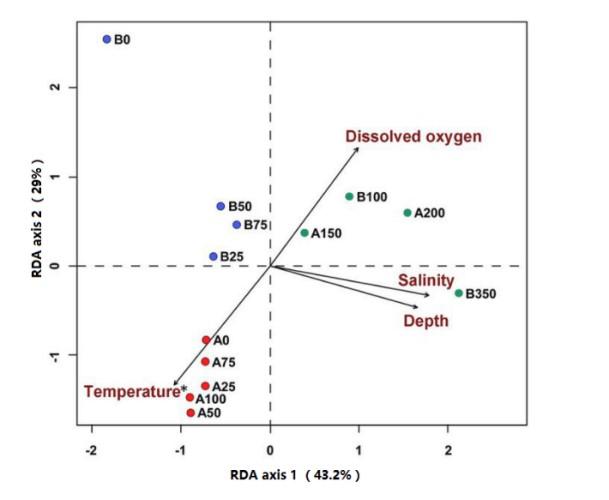
LEfSe分析LEfSe分析主要目的是进行两组或多组之间的比较,找到不同组间在丰度上有显著性差异的物种(biomarker)。
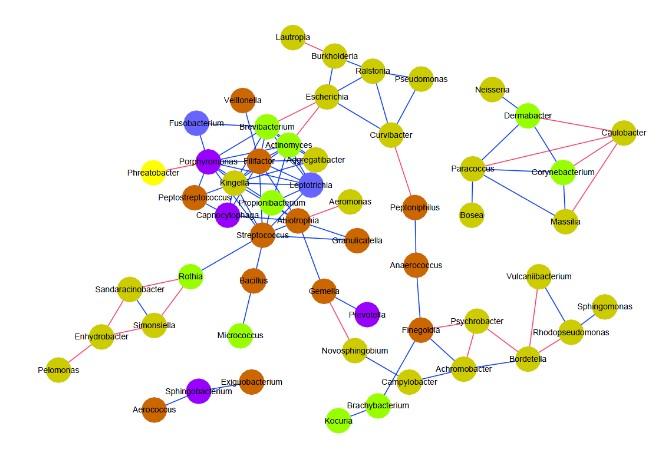
Spearman关联分析主要目的是观察微生物间(或OTU)的相互作用,通过斯皮尔曼(Spearman)关联系数计算等方法,找寻微生物在不同环境下的可能的相互“协作”或“竞争” 的关系。