原核转录组测序是指利用高通量测序技术对原核生物的转录本(mRNA和非编码RNA)进行测序,全面快速地获取特定微生物在特定状态下的所有转录本的信息。通过转录组测序不仅能够对mRNA进行表达定量分析,分析差异表达基因及其相应功能,同时还能够分析Non-coding RNA(sRNAs),揭示微生物不同表型形成的分子调控机制和功能。 由于原核生物mRNA不具有polyA尾结构,需要采用去除rRNA的方法构建文库,联川生物针对不同的研究物种采取有效的方法去除rRNA,保证数据质量。
技术优势
磁珠法去除rRNA,rRNA去除效率好,数据质量高
采用链特异性建库,建库稳定性好
除进行mRNA定量分析外,还可以进行反义转录本预测,基因结构分析等;
可以同时进行mRNA和sRNA分析,sRNA 预测,变异分析等
技术路线
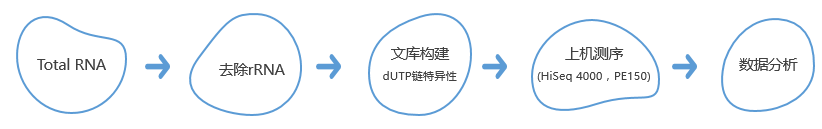
分析内容

样本类型
微生物菌体(≥ 5×107),组织,环境样品,总RNA等
建议总RNA起始量:≥3μg;浓度:≥70 ng/μL。
Q:原核生物与真核生物在进行转录组测序文库构建时有什么区别?A:在原核生物中,mRNA只占全部RNA的1-5%,其余绝大部分是核糖体RNA(rRNA),因此在开展转录组测序前,需先将mRNA纯化。然而,原核生物mRNA不具有polyA结构,无法直接利用oligoT将mRNA纯化出来,如果直接用total RNA进行测序,数据质量将非常差,大部分序列都来自rRNA。
提高原核生物中mRNA的量,最主要的方式是去除total RNA中的rRNA。在建库过程中显加入磁珠将rRNA吸附,过滤掉磁珠后的体系再进行下游建库。
全基因组测序和转录组揭示蓝藻对青藏高原极端气候的适应机制
研究背景
青藏高原不仅是世界上最高且规模最大的高原,同时具有包括极大的温差、低氧浓度、低压、强紫外线辐射以及狂风等在内的极端环境,且有许多独特的环境,包括雪山、盐水湖泊及干旱沙 漠等,具有极高的生物多样性,为研究适应性进化提供了一个理想的天然实验室。蓝藻是最早的光合放氧生物,几乎可以将所有纬度范围的地区作为栖息地,对于全球生态具有相当大的重要 性;蓝藻可以适应可变渗透压、持续低温及强烈的紫外线辐射等环境。
研究结果
青藏高原蓝藻基因组序列是 5.9 Mb,具有 39.2%的 G+C 含量,总共包括5362 个 CDS。系统进化树表明,这一菌株属于 Trichormus 和 Anabaena 集群。T. sp. NMC-1 和6个近 缘种之间的基因组对比显示,在 T. sp. NMC-1 基因组中,功能未知的基因占据了更高的比例(28.12%)。
转录组分析发现表达显著上调的基因参与膜生物起源、翻译、核糖体结构和生物起源、次生代谢产物生物合成、氨基酸运输和代谢、防御机制等过程;相比之下,下调基因主要参与信 号转导机制、膜生物起源及能源生产和转换。进一步的分析表明,CheY-like 基因、胞外多糖和类菌胞素氨基酸样的氨基酸,可能在极端环境的适应性中扮演重要的角色。
研究结论
青藏高原引起极端环境而具有最高的生物多样性,为研究适应性进化提供了一个理想的天然实验室。该研究绘制了青藏高原蓝藻的基因组序列草图,并在低温下进行了全转录组测序,以探讨 T. sp. NMC-1 适应特殊环境的遗传学机制。明显正向选择的、扩张纯正群的和差异表达的一些基因参与信号转导、细胞壁/膜生物起源、次生代谢产物的生物合成、能源生产和转换,旨在阐 述特殊的适应特征。这些结果表明,蓝藻对青藏高原极端环境的适应性背后,有着复杂的遗传学机制。
参考文献
Qin Q, Huang Y, Ji Q, et al. The genome and transcriptome ofTrichormussp. NMC-1: insights into adaptation to extreme environments on the Qinghai-Tibet Plateau:[J]. Scientific Reports, 2016, 6:29404.
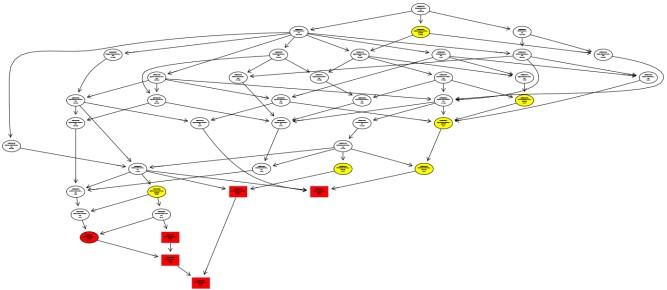
差异基因 GO 富集 DAG 图有向无环图(Directed Acyclic Graph, DAG)为差异基因GO富集分析结果的图形化展示方式。图中的分支代表包含关系,从上至下所定义的功能范围越来越小,一般选取GO富集分析的结果前10位作为有向无环图的主节点,并通过包含关系,将相关联的GO Term一起展示,颜色的深浅代表富集程度。
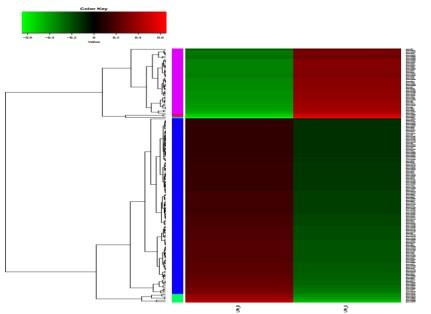
差异基因表达模式聚类将有显著差异的基因/转录本进行表达模式聚类分析,采用距离计算算法: 样本间为spearman,基因间为pearson,采用的聚类方法为hcluster(complete算法)。
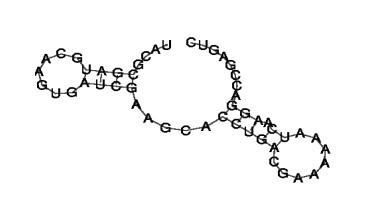
sRNA 预测原核生物中,长度在 50~500nt 的非编码 RNA 通常定义为小 RNA(small RNA, sRNA)。用 Rockhopper 软件 发现新转录本, 通过将其与 Uniport 数据库进行注释,将未注释到数据库的转录本作为潜在的非编码 sRNA。用RNAFold 分析其茎环结构,进行 二级结构预测。