背景简介
细菌基因组重测序是指对基因组序列已知的细菌个体进行基因组测序,通过与已知的参考基因组比对,获得该细菌个体或者群体的差异的测序方法,这些差异主要包括SNP,InDel和SV。目前微生物基因组重测序被广泛应用于病原微生物的检测及鉴定、病原菌演变及起源、致病菌种群结构及种群结构的进化等众多方面。
技术优势
细菌基因组重测序能够发现未知的遗传变异信息。
凭借二代测序,细菌基因组重测序获得数据通量更高,速度更快,成本更低。
细菌基因组重测序可以更全面的检测SNP、InDel、SV等多种遗传变异类型。
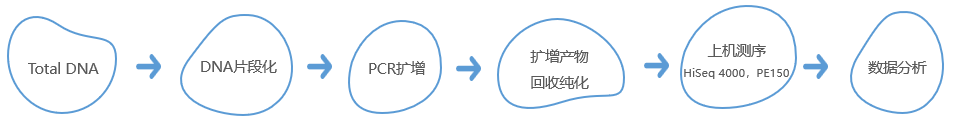
技术路线
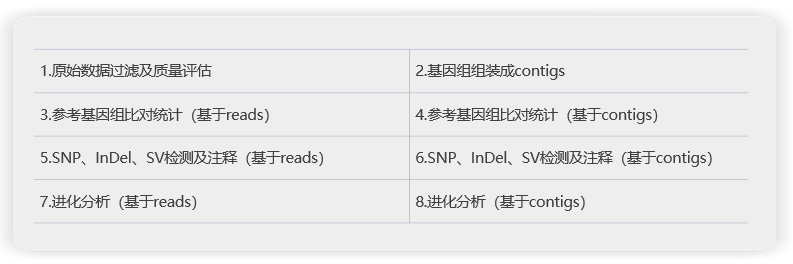
分析内容
样本类型
细菌菌体,环境样品,总DNA等
建议总DNA起始量:≥5 μg,最低起始量:0.5ug,浓度≥30 ng/μl(Qubit定量)
Q1:细菌基因组完成图是否可以组装出质粒?
A:细菌细胞内一般会存在部分质粒,细菌基因组完成图可以组装出部分质粒信息,但是由于建库的长度以及质粒的数量限制,不保证完全组装出所有质粒。
Q 2:细菌基因组测序中比较基因组分析是什么?
A:细菌基因组比较基因组分析,主要是分析有亲缘关系的细菌基因之间的差别,通过比较基因组分析亲缘远近关系,特有和共有基因以及共线性分析等,找到差异基因,对差异基因进行功能注释,解析近缘关系细菌间生理或形态差别的原因。
Q3:在重测序中,为什么只能得到插入/缺失了碱基的数目,却得不到插入/缺失的具体位置和序列信息?如何能够获得具体的序列信息呢?
A:在重测序中,SV检测分析是可以得到样本相对于参考基因组的一个大概的DEL序列的。由于重测序中只是对于文库片段的两端进行测序,所以中间的INS序列暂时无法检测到;理论上,可以对插入位置附件设计引物,通过PCR扩增出具体的序列,此外,也可以通过局部组装附近的reads来获取中间的序列信息(主要取决于局部组装的效果)。
肠外致病性大肠杆菌的大规模基因组测序
研究背景
大肠杆菌(E. coli)为埃希氏菌属(Escherichia)代表菌。一般多为条件致病菌,某些血清型菌株的致病性强,严重腹泻和败血症,统称致病性大肠杆菌。其中,致病性大肠杆菌的抗生素耐药性,是相关医疗实践中的一个重要问题。
方法流程
取材:1. 5个月时间从277个病人尿液中分离出288株尿源E.coli ;2. 3年时间从47名病人血液中分离培养出92株血源E.coli
建库:构建小片段文库
测序:Illumina HiSeq 2000
分析:1. 基因组组装;2. Core-pan基因分析;3. 全基因组关联分析
研究结果
1. Core-pan基因分析
通过Core-pan基因分析发现,ExPEC E.coli具有高度遗传异质性,并区分为不同种系。不同致病因素及抗生素抗性表型对应人体不同部位的感染性。
2. 致病性研究
高分辨率的分子流行病学研究显示,经由血液传播的亚种比例较低,仅占全部亚种的28%。
3. 抗性基因挖掘
全基因组关联分析发现部分抗生素抗性相关的基因,并成功验证了部分已知抗性的基因。
研究结论
本文尝试将大量高通量数据应用到医疗机构,并从中寻找到菌种分类、进化及抗生素抗性等重要基因信息的获得途径,为后续疾病相关大规模微生物全基因组测序提供了范例。
SNP分析SNP全称Single Nucleotide Polymorphisms,是指在基因组上单个核苷酸 的变异,形成的遗传标记,其数量很多,多态性丰富。基因组上单个核 苷酸的变异包括置换,缺失和插入。采用FreeBayes对各样品比对结果进行个体SNP的检测。

InDel分析InDel (Insertion-Deletion) 是指相对于参考基因组,样本中发生的小片段的 插入缺失,该插入缺失可能含一个或多个碱基。根据InDel在基因组中的位 置,可以分为编码区序列的InDel和非编码区序列的InDel。编码序列中的 InDel发生与编码蛋白质的功能和氨基酸位点在结构和功能上的重要性有关。 采用FreeBayes对各样品比对结果进行个体InDel的检测。

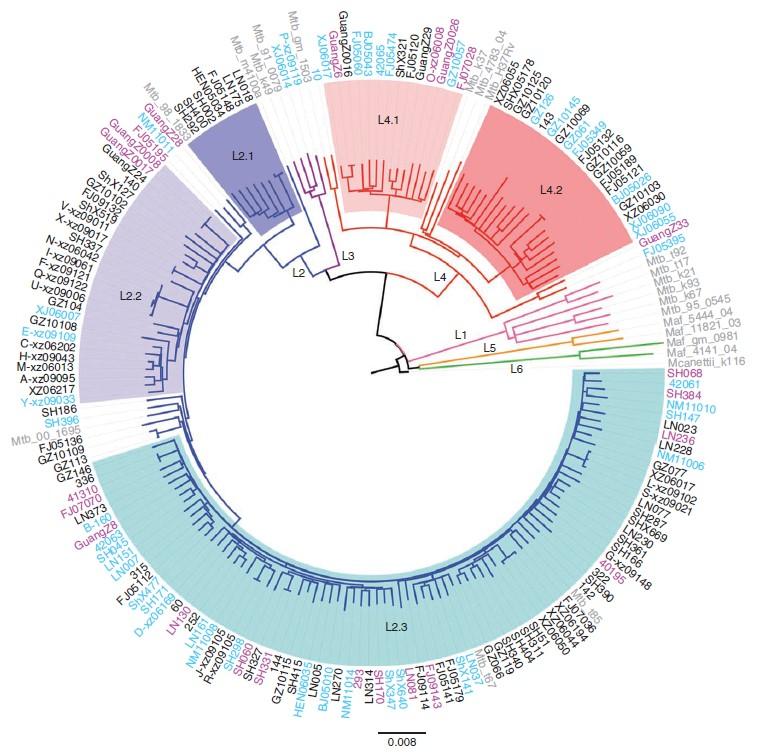
SNP进化树分析在生物学中,进化分析是指根据遗传或者表型的差异研究推断不同物种或者个体间的进化关系的一门学科。进化分析的结果通常用进化树的形式展示,在进化树上每个结点代表一个物种或个体,那么两个结点之间的最短距离就表示相应的两个物种之间的差异程度。根据每个个体检测到的SNP位点,过滤掉彼此距离较近的SNP位点(<20bp) ,因为这些位点可能是由基因组重组产生,并不能真正反映物种间的进化关系。将每个个体非重组的SNP位点碱基串联成一条序列,得到个体多序列比对的结果,最后构建最大似然进化树。